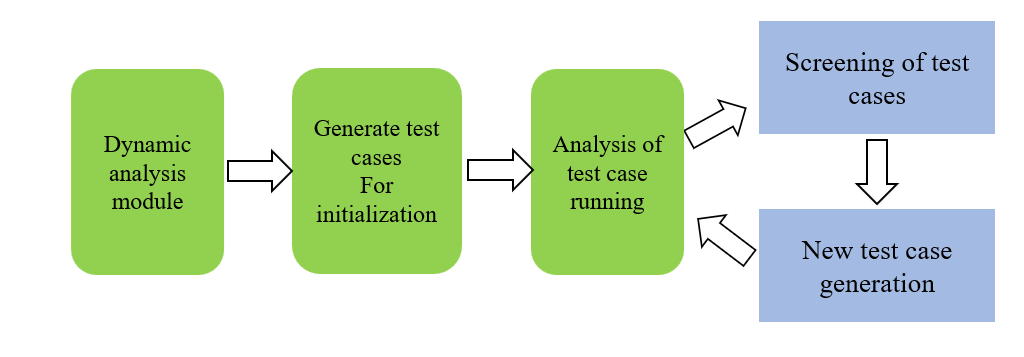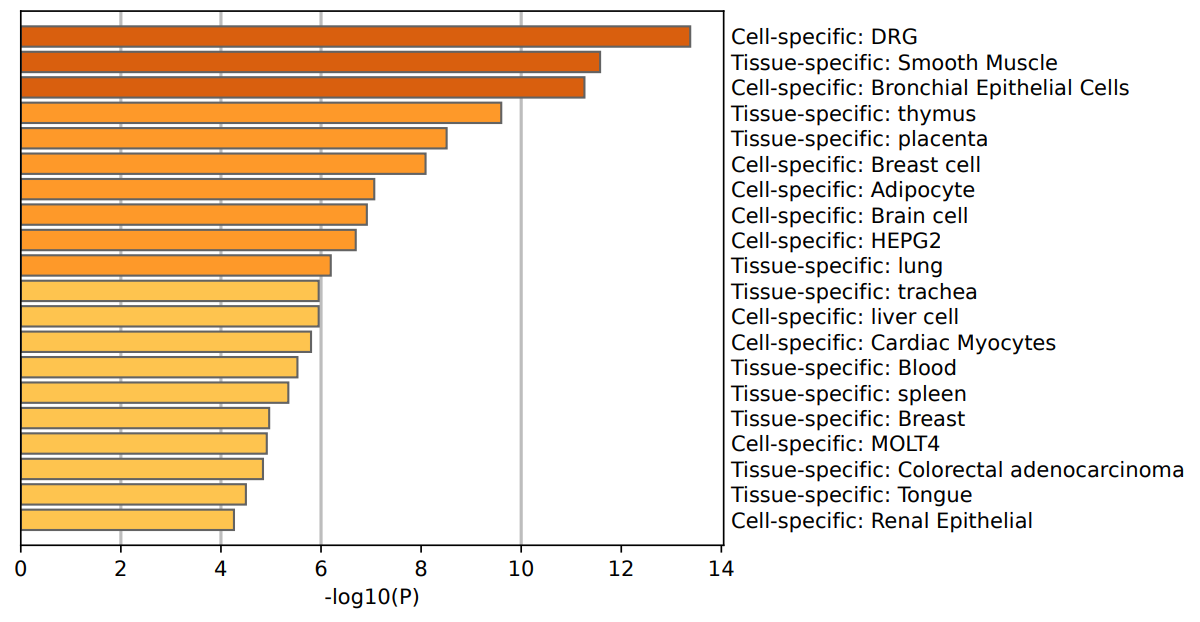
Breast cancer is one of the most common malignant tumors among women globally, posing significant threats to women's health and lives. Identifying specific molecular markers is crucial for early diagnosis, precision treatment, and accurate prognostic assessment of breast cancer. In this study, spatial transcriptomics technology combined with machine learning methods successfully identified molecular markers capable of effectively distinguishing invasive ductal carcinoma (IDC) from ductal carcinoma in situ (DCIS) and lobular carcinoma in situ (LCIS). By analyzing the gene expression profiles of 36,601 genes across 3,798 cells, significant differentially expressed genes (DEGs) were screened using the DEsingle method. Functional enrichment analysis indicated that these genes are significantly associated with breast cancer-related pathways, breast cell-specific expression, and the regulation of core transcription factors, such as TP53, SP1, and NFKB1. Further classification analysis employing machine learning models including random forest, decision tree, support vector machine, and logistic regression revealed that the random forest model demonstrated the highest performance, achieving an accuracy rate of 95.78%. Ultimately, ten key molecular markers were identified: MGP, ALB, S100G, KRT37, SERPINA3, AC087379.2, ZNF350-AS1, IGHG3, IGHG4, and IGKC. These markers exhibited robust discrimination between IDC and DCIS/LCIS, suggesting their potential roles in tumor invasion and metastasis. This study provides novel molecular evidence for early diagnosis, individualized treatment, and prognostic evaluation of breast cancer, contributing new research insights and theoretical support for precision medicine approaches in breast cancer.